

Implementation and preparation of risk management file within the scope of MDR (2017/745).
Risk Management and MDR
Risk Management Regulatory
To ensure that devices manufactured in series production continue to be in conformity with the requirements of Medical Device Regulation 2017/747 (MDR) and that experience from the use of the devices they manufacture is taken into account for the production process, all manufacturers should have a quality management system and a post-market surveillance system in place which should be proportionate to the risk class and the type of the device in question according to MDR, Preface, Section 32. In addition, in order to minimize risks or prevent incidents related to devices, manufacturers should establish a system for risk management and a system for reporting of incidents and field safety corrective actions.
The risk management system should be carefully aligned with and reflected in the clinical evaluation for the device, including the clinical risks to be addressed as part of clinical investigations, clinical evaluation and post-market clinical follow up. The risk management and clinical evaluation processes should be inter-dependent and should be regularly updated (MDR, Foreword Section 33).
Annex I, Chapter 1 of the MDR lists the general requirements regarding the risks of a medical device. Among other things, it states that manufacturers must introduce, implement, document and update a risk management system. Risk management is to be understood as a continuous iterative process during the entire life cycle of a product, which requires regular systematic updating. In implementing risk management, manufacturers must, among other things (MDR, Annex I, Chapter 1, Paragraph 3):
- establish and document a risk management plan for each device;
- identify and analyse the known and foreseeable hazards associated with each device;
- estimate and evaluate the risks associated with, and occurring during, the intended use and during reasonably foreseeable misuse;
- evaluate the impact of information from the production phase and, in particular, from the post-market surveillance system, on hazards and the frequency of occurrence thereof, on estimates of their associated risks, as well as on the overall risk, benefit-risk ratio and risk acceptability
For the establishment of risk management, manufacturers should follow the harmonized standards for this purpose. The MDR, Foreword section states in this regard that given the important role of standardization in the field of medical devices, manufacturers should be able to demonstrate conformity with the essential safety, performance and other regulatory requirements set out in this Regulation, for example on quality and risk management, by complying with the harmonized standards set out in Regulation (EU) No 1025/2012 of the European Parliament and of the Council (15).
For risk management, this would be EN ISO 14971, even though this standard is not yet listed as a harmonized standard for MDR. However, the EU has published a list of standards containing those standards which it intends to harmonize. This also includes EN ISO 14971. (Link: https://ec.europa.eu/docsroom/documents/43584?locale=en)
Risk Management Process
EN ISO 14971 states that a manufacturer shall establish, implement, document and maintain a process to:
- Identify the hazards and hazardous situations associated with the product;
- Assess and evaluate the associated risks;
- Control these risks;
- Monitor the effectiveness of the risk control measures.
The risk management process can be part of a quality management system, e. g. based on ISO 13485:2016. However, this is not required by EN ISO 14971.
The risk management process must include risk analysis, risk evaluation, risk control and activities / data and information during manufacturing and downstream phases of production.
In addition to EN ISO 14971, the Technical Report ISO/TR 24971 can be used as a guideline.
Risk Management Plan (RM Plan)
The risk management plan describes the scope of risk management activities, the responsibilities and authorities of those involved, the criteria for risk acceptability, the production and post-production information to be collected and reviewed for the medical device. Furthermore, the RM plan contains all risk management activities that are carried out during the entire product life cycle. The RM plan is reviewed throughout the life cycle of the device and updated as new information becomes available. In doing so, the scope of activities should be appropriate to the risk associated with the medical device.
Risk Analyses
The risk analysis process consists of the description of the intended use of the medical device and reasonably foreseeable misuse, the identification of the characteristics of the medical device that are related to safety, the identification of hazards and hazardous situations associated with the medical device and the estimation of risks for each hazardous situation.
According to ISO/TR 24971 a hazard is a potential source of a harm. Depending on the specific situation, hazards can have different origins/natures such as electricity, moving parts, infectious bacteria and virus, chemicals and sharp edges. Medical devices cause harm only when a sequence of events occurs that leads to a hazardous situation that then causes harm or results in harm. The sequence of events can be a chronological series of causes and effects as well as combinations of simultaneous events. However, dangerous situations can also occur when there are no faults, e. g. in the normal state of the medical device when it is functioning as intended.
For each of these identified hazardous situations, the manufacturer must assess the associated risk(s). The risk assessment includes an analysis of the probability of occurrence of damage and the severity of the damage (EN ISO 14971). Risk assessment can be performed qualitatively or quantitatively. Several methods can be used for this purpose, such as PHA, FMEA and FTA.
The risk analysis therefore consists of the identification of possible hazards from the medical device and the estimation of probabilities, severity and thus the risks. In order to be able to assess the risks, the manufacturer should determine in advance which risks are considered acceptable and which are considered unacceptable. This is too usually done in the form of a risk assessment matrix (risk acceptance matrix). Examples of risk assessment matrices or diagrams can be found, for example, in ISO/TR24971.
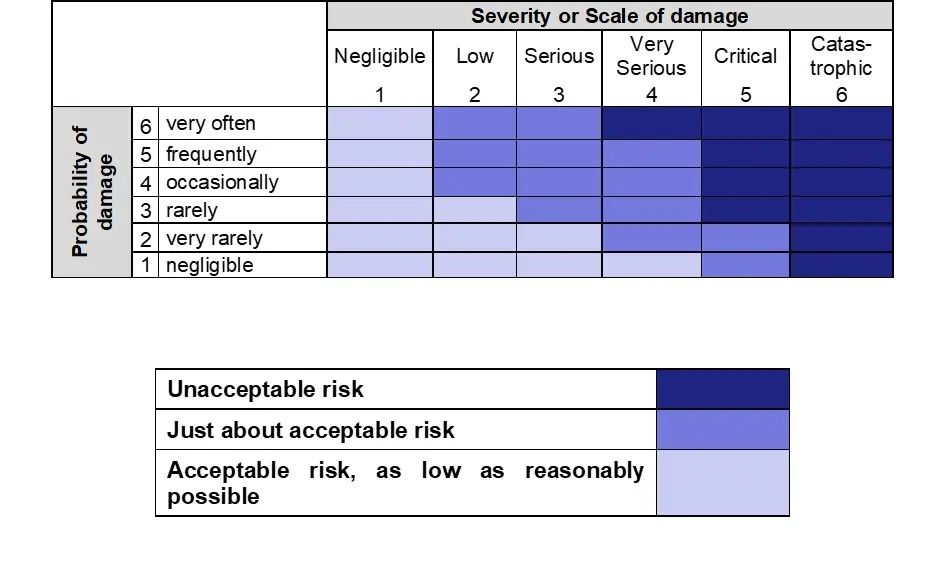
Specific criteria may be established for each type of medical device (or family of medical devices), depending on their characteristics and intended use, or the same criteria may apply to all medical devices. The criteria for the acceptance of risks are recorded in the risk management plan.
Risk Evaluation
During risk evaluation, the manufacturer compares the assessed risks with the acceptance criteria for the acceptability of risks previously defined in the risk management plan and determines whether or not these criteria are met.
Risk Control
If it is determined during the risk analysis that there are unacceptable risks, the manufacturer must minimize these risks. In general, the benefits of the medical device must outweigh the risks, so the risks must be controlled.
In minimizing risk, ISO EN 14971 requires the manufacturer to proceed according to the following sequence:
- Inherently safe design and safe manufacturing;
- Protective measures in the medical device itself or in the manufacturing process;
- Information for safety and, where applicable, training of users.
In this context, it applies that the severity of the damage and / or the probability of damage can be reduced if measures are taken to control the risk. Explanation of the individual points mentioned above are contained in ISO/TR 24971. The risk mitigation measures shall be documented in the risk management file.
If, during the analysis of the risk control options, the manufacturer determines that the mitigation of the risk is not feasible, the manufacturer must perform a benefit-risk analysis for the residual risk (EN ISO 14971). Subsequently, the measures must be taken and the effectiveness of the measures taken must be verified. The results of this verification must in turn be documented in the RM file.
Residual Risks Evaluation
After the risk minimization measures have been implemented, the manufacturer must review the residual risks again. According to EN ISO 14971, residual risks are the risks that remain after the risk minimization measures have been implemented. The acceptance criteria established in the risk management plan are again used for this purpose. The residual risk is again either acceptable or unacceptable. If it is determined to be unacceptable, further risk control options should be explored. If further risk control is not feasible, a benefit-risk analysis can be performed.
It is also important to note that the risk control measures taken may give rise to further risks, which must then again be evaluated against the established acceptance criteria and may give rise to further risk mitigation measures.
Benefit-Risk Analyses
ISO 14971:2019 allows the manufacturer to perform a risk-benefit analysis for those risks that are judged unacceptable using the criteria established in the risk management plan and for which further risk control is not practicable. If a residual risk is judged to be unacceptable using the criteria established in the risk management plan and further risk control measures are not feasible, the manufacturer may compile and evaluate data and literature (e.g., data on equivalent and similar products in the marketplace, state of the art, etc.) to determine whether the benefits of the intended use outweigh this residual risk.
Evaluation of the overall residual risk
ISO EN 14971 requires that the overall residual risk is evaluated in relation to the benefit of the intended use of the medical device. For this purpose, both the acceptance criteria of the overall residual risk and the method for assessing the overall residual risk must be included in the risk management plan.
The evaluation of the overall residual risk is the point at which the residual risk is considered from a comprehensive perspective. All identified hazardous situations have been evaluated and all risks have been reduced to an acceptable level or accepted based on a risk-benefit analysis. Now the manufacturer assesses whether the overall residual risk associated with the medical device meets the criteria for acceptability of the overall residual risk. The evaluation may conclude that the medical device is safe. If the overall residual risk is too high in relation to the benefit, the manufacturer may take further risk minimization measures or make changes to the medical device or its intended purpose.
Risk Management Report / Review (RM-Report)
ISO EN 14971 requires that the final results of the risk management process be reviewed to ensure that the risk management plan has been properly executed, that the overall residual risk is acceptable, and that appropriate methods are in place to collect and review relevant production and post-production information. The risk management review will occur after the implementation and is also a review of all risk control measures control measures, but prior to the commercial release of the medical device. The results of this review are listed in the Risk Management Report. If in e. g. production phase or the downstream phases new information becomes available that affects the safety of the medical device, it may be necessary to update the RM report.
We are happy to support you in this, please contact us!
Source:
Risk Management
Risk management is a process for identifying the hazards and hazardous situations associated with your medical device; for assessing and evaluating the associated risks and controlling these risks; and for monitoring the effectiveness of the risk control measures. In doing so, we can, amongst other, support you in the following ways:
- Create / review / update risk management plan, establish acceptance criteria.
- Database research: Incidents (FDA MAUDE and TPLC, BfArM, EUDAMED (future), etc.)
- Performing / updating / reviewing / supporting the risk analysis
- Create and update the risk management report
With our help you can:
- Avoid unnecessary work.
- Regularly adapt your risk analysis to new risks as they arise.
- Save time and money by having the time-consuming research work bundled and evaluated in one place!
- We prepare your documents quickly and reliably in German or English, using either your own or AnCura's own standard-compliant forms.
- Together we will find solutions, your satisfaction is our top priority!
Generally, we offer our services in German as well as in English.
If we have aroused your interest, please contact us. Call us, write to us or fill out our contact form. We look forward to hearing from you.